Prerank算法
用途与运行方式
场景一:对SDAS DEG分析得到的所有差异基因进行prerank分析
SDAS geneSetEnrichment prerank \ -i de_t-test.anno_rctd.SmoothMuscle-vs-Endo.all.csv -o ./ \ --species human场景二:只用感兴趣的数据库进行分析
SDAS geneSetEnrichment prerank \ -i de_t-test.anno_rctd.SmoothMuscle-vs-Endo.all.csv -o ./ \ --gmt sdas_deg_enrichment/lib/GSEADB/h.all.v2024.1.Hs.symbols.gmt,sdas_deg_enrichment/lib/GSEADB/KEGG_2021_Human.gmt场景三:只对感兴趣的通路进行作图,将感兴趣的通路全名写入一个txt文档里面,每个通路一行,然后将这个txt文档通过
--pathways参数传入分析流程。需要注意的是使用的数据库中必须包含这些指定的通路名称。SDAS geneSetEnrichment prerank \ -i de_t-test.anno_rctd.SmoothMuscle-vs-Endo.all.csv -o ./ \ --gmt sdas_deg_enrichment/lib/GSEADB/h.all.v2024.1.Hs.symbols.gmt,sdas_deg_enrichment/lib/GSEADB/KEGG_2021_Human.gmt \ --pathwas ./term.txt
输入参数说明
| prerank参数 | 是否必须 | 默认值 | 描述 |
|---|---|---|---|
| -i / --input | 是 | SDAS DEG分析得到的all.csv文件 | |
| -o / --output | 是 | 结果存放路径 | |
| --species | 否 | human | 指定或构建好的物种的数据库,'human' 或 'mouse',默认 'human',当指定--gmt参数时,该参数不起作用 |
| --gmt | 否 | gmt格式的数据库文件,其中gene name信息必须为大写,多个文件时用','隔开 | |
| --graph | 否 | 10 | 筛选top数量的通路进行画图,默认'10',设置了--pathways参数时,该参数不起作用 |
| --pathways | 否 | 通过txt文件指定1到多个感兴趣的通路进行画图 | |
| --min_size | 否 | 15 | 基因集中包含的输入基因最小数量。默认:15 |
| --max_size | 否 | 20000 | 基因集中包含的输入基因最大数量。默认:20000 |
| --label | 否 | 表型标签参数需要定义两个参数。默认:('Pos','Neg') | |
| -v / --verbose | 否 | 启用详细模式,打印任务进度。默认:False | |
| --permu_num | 否 | 1000 | 随机置换次数(用于计算esnulls)。默认:1000 |
| --weight | 否 | 1 | 排序指标权重(用于调整输入基因权重),可选值:{0, 1, 1.5, 2}。默认:1 |
| --ascending | 否 | 设置排序指标为升序(若指定此参数则ascending=True)。默认:False(降序) | |
| --seed | 否 | 123 | 随机数种子。默认:123 |
| --threads | 否 | 1 | 并行计算使用的线程数。默认:1 |
输出结果展示
| prerank结果文件 | 描述 |
|---|---|
prerank_{database}.csv |
csv格式的结果文件 |
prerank_{database}:top10.pdf/png |
pdf和png格式的图像文件 |
- csv文件格式:
prerank_{database}.csv,文件结果跟gsea类似,包含Name,Term,ES,NES,NOM p-val,FDR q-val,FWER p-val,Tag %,Gene %,Lead_genes这几列,其中Term是通路名称;ES是富集得分(Enrichment Score),反映基因集成员在排序基因列表(如差异表达基因排序)中的富集程度,正ES:基因集在排序列表顶部富集(与表型正相关),负ES:基因集在排序列表底部富集(与表型负相关);NES是标准化富集得分 (Normalized Enrichment Score);NOM p-val是名义p值;FDR q-val是校正后的p值;FWER p-val是族系错误率校正后的p值;Tag %是基因集中位于排序列表核心富集区域的基因百分比;Gene %是分析中实际使用到的基因占基因集总基因数的百分比;Lead_genes是对富集得分(ES)贡献最大的核心基因。
| Name | Term | ES | NES | NOM p-val | FDR q-val | FWER p-val | Tag % | Gene % | Lead_genes |
|---|---|---|---|---|---|---|---|---|---|
| prerank | HALLMARK_MYC_TARGETS_V1 | 0.7472938191195556 | 2.39333105644001 | 0.0 | 0.0 | 0.0 | 160/195 | 18.89% | RPL14;HNRNPA2B1;... |
| prerank | HALLMARK_OXIDATIVE_PHOSPHORYLATION | 0.7431758291176868 | 2.376055485647371 | 0.0 | 0.0 | 0.0 | 168/200 | 20.44% | MDH2;COX8A;... |
| prerank | HALLMARK_ALLOGRAFT_REJECTION | 0.744882727767552 | 2.3688992213810462 | 0.0 | 0.0 | 0.0 | 118/194 | 14.03% | ITGB2;HLA-DRA;... |
| prerank | ... | ... | ... | ... | ... | ... | ... | ... | ... |
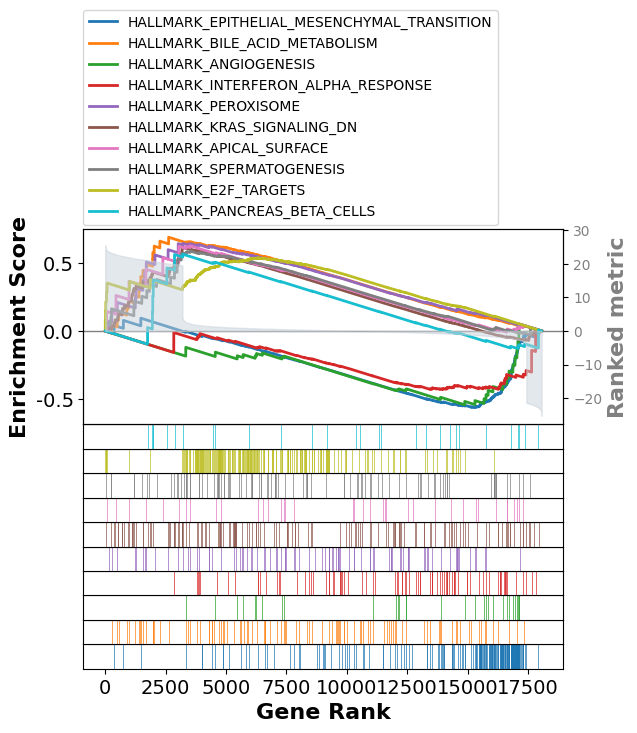
- top Terms富集曲线图:
prerank_{database}:top10.pdf/png(见下图示例),图中Enrichment Score(ES)的正负直接反映基因集在基于log2FC排序基因列表中的分布模式:ES为正,表示基因集成员集中在排序列表的顶部,基因集与表型正相关;ES为负,表示基因集成员集中在排序列表的底部,基因集与表型负相关。