单细胞/pseudobulk差异分析
用途与运行方式
1. 单细胞水平差异分析(无生物学重复)
支持t-test、wilcoxon、MAST
场景一:指定类别1与类别2差异分析
SDAS DEG -i st.h5ad -o outdir --group_key leiden --de_method wilcoxon \ --ident1 1 --ident2 2 \ --fdr 0.05 --log2fc 1场景二:每个类别与其余类别分别做差异分析
SDAS DEG -i st.h5ad -o outdir --group_key leiden --de_method wilcoxon \ --fdr 0.05 --log2fc 1场景三:对obs的某列做子集后再差异分析
SDAS DEG -i st.h5ad -o outdir --group_key leiden --de_method wilcoxon \ --ident1 1 --ident2 2 \ --fdr 0.05 --log2fc 1 \ --subset_key cell_type --subset_values B
2. pseudobulk差异分析(有生物学重复)
推荐DESeq2、edgeR(pseudobulk分析),需指定--sample_key,且每组样本数需满足方法要求(DESeq2≥3,edgeR≥2)
场景一:两组样本直接差异分析
SDAS DEG -i st.h5ad -o outdir --group_key sampleID --de_method DESeq2 \ --ident1 Tumor --ident2 Normal \ --fdr 0.05 --log2fc 1 \ --sample_key sampleID场景二:子集后做pseudobulk差异分析
SDAS DEG -i st.h5ad -o outdir --group_key sampleID --de_method DESeq2 \ --ident1 Tumor --ident2 Normal \ --fdr 0.05 --log2fc 1 \ --sample_key sampleID \ --subset_key cell_type --subset_values B
输入参数说明
| DEG参数 | 是否必须 | 默认值 | 描述 |
|---|---|---|---|
| -i / --input | 是 | Stereo-seq h5ad,要求原始矩阵 | |
| -o / --output | 是 | 分析结果存放路径 | |
| --de_method | 是 | 指定差异分析方法:{t-test,wilcoxon,MAST,DESeq2,edgeR} | |
| --group_key | 是 | 需要进行差异分析的对象所在的obs名称 | |
| --ident1 | 否 | 用于分析差异表达基因的对象,类似处理组,不写则按1-vs-rest方式逐个输出 | |
| --ident2 | 否 | 用于做对照的对象,类似对照组,不写则将剩余元素当作对照,类似1-vs-rest | |
| --sample_key | 否 | 存放样本信息的obs名称,使用DESeq2或edgeR时进行pseudobulk差异分析时必须指定且必须存在生物学重复样本,使用DESeq2时必须有>=3个生物学重复,使用edgeR时必须有>=2个生物学重复 | |
| --subset_key | 否 | 需要提取的值所属的分组列名称 | |
| --subset_values | 否 | 需要提取的值,存在多个时用','分隔 | |
| --layer | 否 | 指定表达矩阵,不指定时用adata.raw.X或adata.X | |
| --gene_symbol_key | 否 | real_gene_name | Stereo-seq h5ad.var中表示基因名(symbol)的列的名称 (_index 表示使用h5ad.var.index) |
| --fdr | 否 | 0.05 | Padj(FDR)的阈值,用于筛选显著差异基因 |
| --log2fc | 否 | 1 | log2FC的绝对值阈值,用于筛选显著差异基因 |
| --genelist | 否 | 5 | 在图中标出感兴趣的基因,多个基因可用','分隔,默认用显著上调和下调各5个基因 |
| --add_label | 否 | 对h5ad的obs增加额外的标签 | |
| --min_gene | 否 | 0 | 一个bin/cell允许的最少基因数 |
| --max_gene | 否 | 一个bin/cell允许的最多基因数 | |
| --min_cell | 否 | 0 | 某个基因最少存在于多少个bin/cell中 |
输出结果展示
| 结果文件 | 描述 |
|---|---|
de_{method}.{group_key}.{ident1}-vs-{ident2}.raw.csv |
软件原始的输出结果 |
de_{method}.{group_key}.{ident1}-vs-{ident2}.all.csv |
从原始结果中提取出geneID、log2FC、Pvalue、FDR等信息的结果 |
de_{method}.{group_key}.{ident1}-vs-{ident2}.sig_filtered.csv |
根据log2FC和Pvalue过滤后的显著差异结果 |
de_{method}.{group_key}.{ident1}-vs-{ident2}.png/pdf |
png或pdf格式的火山图 |
- raw文件格式示例:
de_{method}.{group_key}.{ident1}-vs-{ident2}.raw.csv这个文件是差异分析软件生成的分析结果,里面可能包含除了基因名、差异倍数、Pvalue、adjusted Pvalue(FDR)等信息外的其他信息。
| names | scores | logfoldchanges | pvals | pvals_adj |
|---|---|---|---|---|
| MTATP6P1 | 16.74336 | 1.3794351 | 1.3877341418899603e-42 | 2.2785333033340524e-39 |
| AGR2 | 13.671169 | 1.7758344 | 1.419568544127444e-32 | 1.1147316293689464e-29 |
| CLDN4 | 13.663365 | 1.9820584 | 1.9626883546881656e-34 | 1.6880054463820458e-31 |
| ... | ... | ... | ... | ... |
- all/sig_filtered文件格式示例:
de_{method}.{group_key}.{ident1}-vs-{ident2}.all.csv文件是从差异分析软件生成的分析结果中提取了基因名、差异倍数、Pvalue、adjusted Pvalue(FDR)重新统一命名后的结果文件,de_{method}.{group_key}.{ident1}-vs-{ident2}.sig_filtered.csv文件是根据log2FC和FDR阈值进行筛选后得到的显著差异基因列表。
| gene | log2FC | pvalue | FDR |
|---|---|---|---|
| MTATP6P1 | 1.3794351 | 1.3877341418899603e-42 | 2.2785333033340524e-39 |
| AGR2 | 1.7758344 | 1.419568544127444e-32 | 1.1147316293689464e-29 |
| CLDN4 | 1.9820584 | 1.9626883546881656e-34 | 1.6880054463820458e-31 |
| ... | ... | ... | ... |
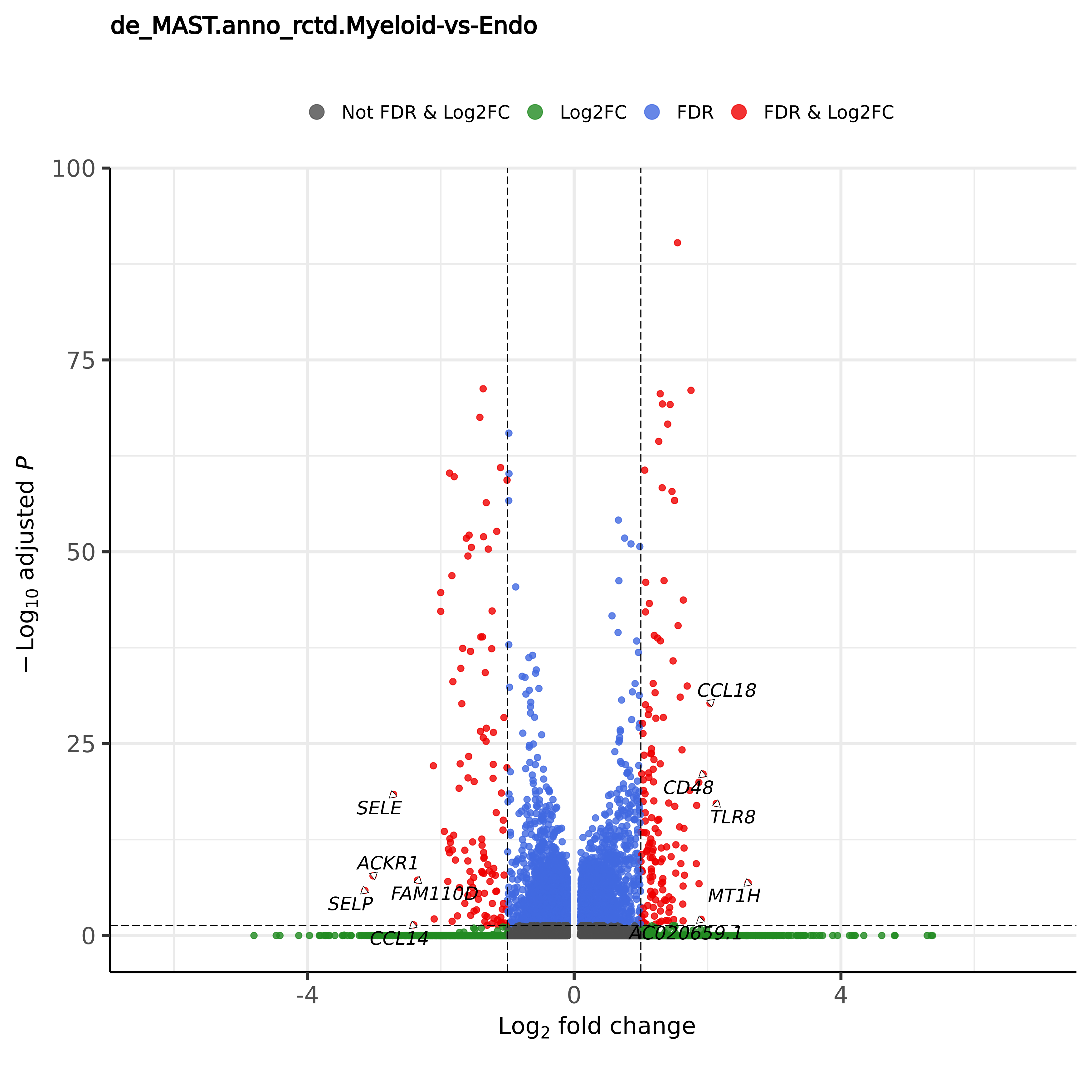
- 火山图结果示例:
de_{method}.{group_key}.{ident1}-vs-{ident2}.png/pdf图中红色的点表示同时达到log2FC和FDR筛选阈值的显著差异基因,蓝色的点表示达到FDR筛选条件但没达到log2FC条件的基因,绿色的点表示达到log2FC筛选条件但没达到FDR条件的基因,同时,默认会在图中标出差异倍数最高和最低的5个基因,如果需要在图中标注指定的基因,可以通过genelist参数进行指定(例如, --genelist geneA,geneB,geneC)。
结果解读说明
- 基因名唯一化
- 差异分析前会自动对基因名进行
make_unique,所有输出和作图均使用唯一化后的基因名。
- 差异分析前会自动对基因名进行
细胞和基因过滤
- 支持通过
--min_gene、--max_gene、--min_cell等参数对细胞和基因进行过滤。若h5ad已过滤,可不再设置。
- 支持通过
调参建议
- 超过200k的bin/cell数据量时MAST无法成功运行,这种情况下可以通过设置更严格的过滤参数(
min_gene和min_cell)减少bin/cell的数量再进行分析。