Hotspot算法
用途
使用Hotspot算法进行空间基因共表达基因集识别
运行方式
SDAS coexpress hotspot -i st.h5ad -o outdir --bin_size 100 \
--layer raw_counts \
--selected_genes top5000 \
--moran_path ./moran.csv \
--n_cpus 8 \
--seed 42 \
--fdr_cutoff 0.05 \
--model bernoulli
输入参数说明
| 参数 | 是否必须 | 默认值 | 描述 |
|---|---|---|---|
| -i / --input | 是 | Stereo-seq h5ad,要求有原始表达矩阵 | |
| -o / --output | 是 | 输出文件夹 | |
| --bin_size | 是 | 50 | 分辨率Bin大小(20, 50, 100, 200, cellbin),与输入h5ad一致,画图与计算均需要 |
| --layer | 否 | 指定h5ad中原始表达矩阵的layer层 (例如layers[‘raw_counts’] | |
| --selected_genes | 否 | top5000 | 基因列表(topn高变基因, full全部基因) |
| --moran_path | 否 | 已计算好的基因莫兰指数列表路径 | |
| --n_cpus | 否 | 8 | 并行计算进程数 |
| --seed | 否 | 42 | 随机种子 |
| --fdr_cutoff | 否 | 0.05 | 统计检验空间高变基因与共表达基因集的FDR矫正阈值 |
| --model | 否 | normal | 统计检验假设(normal, bernoulli, danb, none) |
输出结果展示
| 结果文件 | 描述 |
|---|---|
<input_name>_hotspot.module.csv |
空间高变基因(gene symbol+gene id)对应的共表达基因集(module)的共表达基因集的结果csv |
<input_name>_hotspot.h5ad |
含有共表达基因集结果的h5ad文件(adata.obsm[‘module_score_hotspot’]) |
<input_name>_hotspot_module_score_hotspot.png/pdf |
共表达基因集的基因集打分空间热图 |
<input_name>_hotspot.all_coex_heatmap.png/pdf |
共表达基因集的相似性热图 |
<input_name>_hotspot.moran.csv |
如果使用topn计算,输出全部基因的莫兰指数以及P值 |
- 共表达基因集的结果csv:
<input_name>_hotspot.module.csv,以逗号分隔。Hotspot识别的空间高变基因对应的共表达基因集(module)
| geneid | real_gene_name | FDR | Module |
|---|---|---|---|
| ENSG00000163209 | SPRR3 | 0.0 | Module-1 |
| ENSG00000151632 | AKR1C2 | 0.0 | Module-1 |
| ENSG00000170345 | FOS | 0.0 | Module-1 |
| ENSG00000164433 | FABP5 | 0.0 | Module-1 |
| ENSG00000120129 | DUSP1 | 0.0 | Module-1 |
- 共表达基因集的基因集打分空间热图
<input_name>_hotspot_module_score_hotspot.png/pdf:可视化所有共表达基因集(Module)的空间分布模式。图中颜色强度表示共表达基因集表达量的高低
.png)
- 共表达基因集的相似性热图
<input_name>_hotspot.all_coex_heatmap.png/pdf:展示不同共表达基因集(Module)之间的相似性聚类关系。图中颜色表示不同共表达基因集的相似度,红色为高度相似
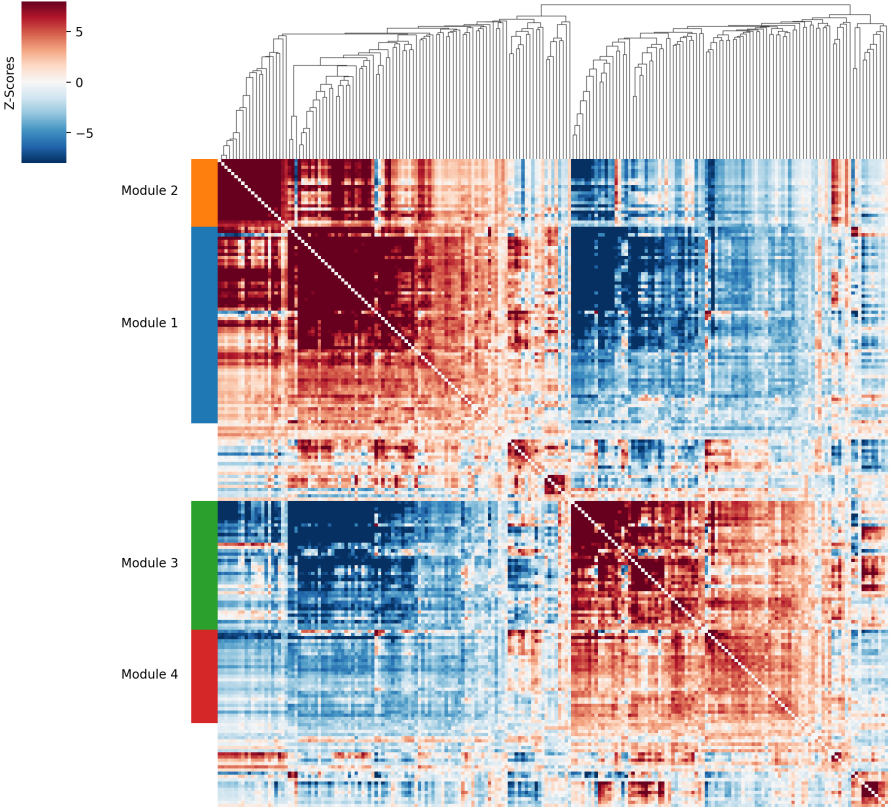
结果解读说明
- 共表达基因集从Module1开始,Module-1/没有Module为不符合共表达基因集聚类要求的基因。
调参建议
- 若样本bin20/50基因数低于200,或其他特殊样本,识别的空间高变基因/共表达基因集较少,建议将
model参数从normal改为bernoulli,并将fdr_cutoff设置为0.05。